



Highlights
Latest Updates
Repeat Proteins Challenge the Concept of Structural Domains
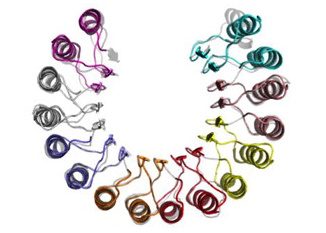
Structural domains are believed to be modules within proteins that can fold and function independently. Some proteins show tandem repetitions of apparent modular structure that do not fold independently, but rather co-operate in stabilizing structural forms that comprise several repeat-units. For many natural repeat-proteins, it has been shown that weak energetic links between repeats lead to the breakdown of co-operativity and the appearance of folding sub-domains within an apparently regular repeat array. The quasi-1D architecture of repeat-proteins is crucial in detailing how the local energetic balances can modulate the folding dynamics of these proteins, which can be related to the physiological behaviour of these ubiquitous biological systems.
Structure-Based Characterization of Multiprotein Complexes |  |
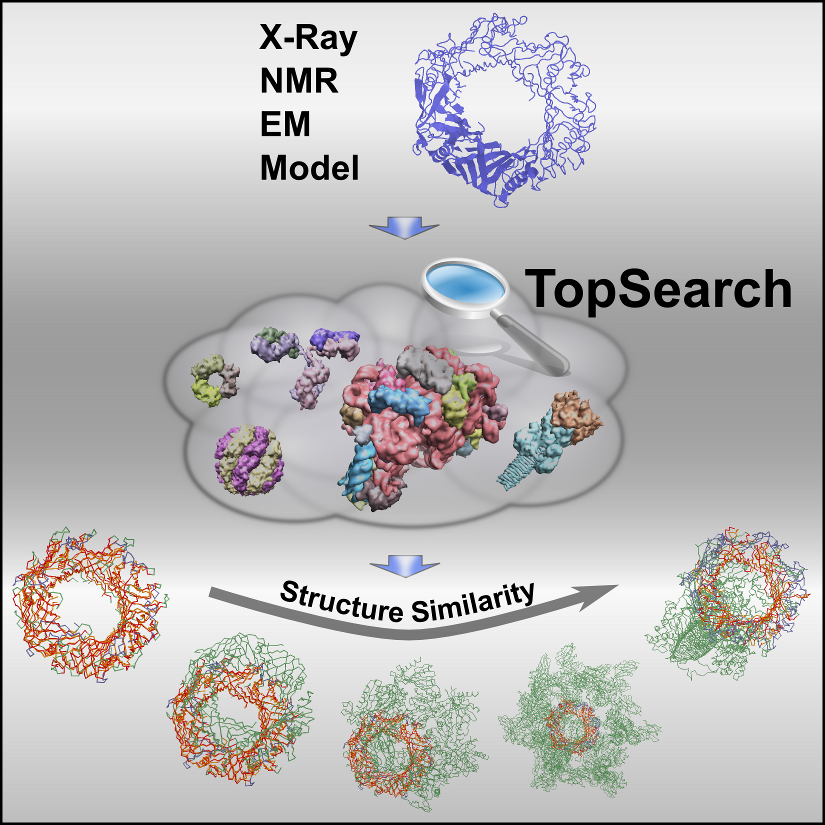
Multiprotein complexes govern virtually all cellular processes. Their 3D structures provide important clues to their biological roles, especially through structural correlations among protein molecules and complexes. The detection of such correlations generally requires comprehensive searches in databases of known protein structures by means of appropriate structure-matching techniques. Here, we present a high-speed structure search engine capable of instantly matching large protein oligomers against the complete and up-to-date database of biologically functional assemblies of protein molecules. We use this tool to reveal unseen structural correlations on the level of protein quaternary structure and demonstrate its general usefulness for efficiently exploring complex structural relationships among known protein assemblies.
![]() Availability: http://topsearch.services.came.sbg.ac.at
Availability: http://topsearch.services.came.sbg.ac.at
Towards the Development of Standardized Methods for Comparison, Ranking and Evaluation of Structure Alignments
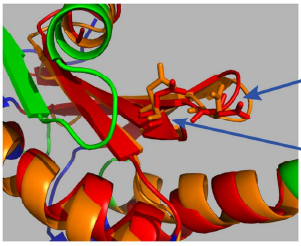
Here we report on the development and application of a new approach for the evaluation of structure alignment results. The method uses the translation vector and rotation matrix to generate the superposition of two structures but discards the alignment reported by the individual programs. The optimal alignment is then generated in standardized form based on a suitably implemented dynamic programming algorithm where the length of the alignment is the single most informative parameter. We demonstrate that some of the most popular programs in protein structure research differ considerably in their overall performance. In particular, each of the programs investigated here produced in at least in one case the best and the worst alignment compared to all others. Hence, at the current state of development of structure comparison techniques, it is advisable to use several programs in parallel and to choose the optimal alignment in the way reported here.
![]() Availability: http://melolab.org/stovca
Availability: http://melolab.org/stovca
Detection of Spatial Correlations in Protein Structures and Molecular Complexes
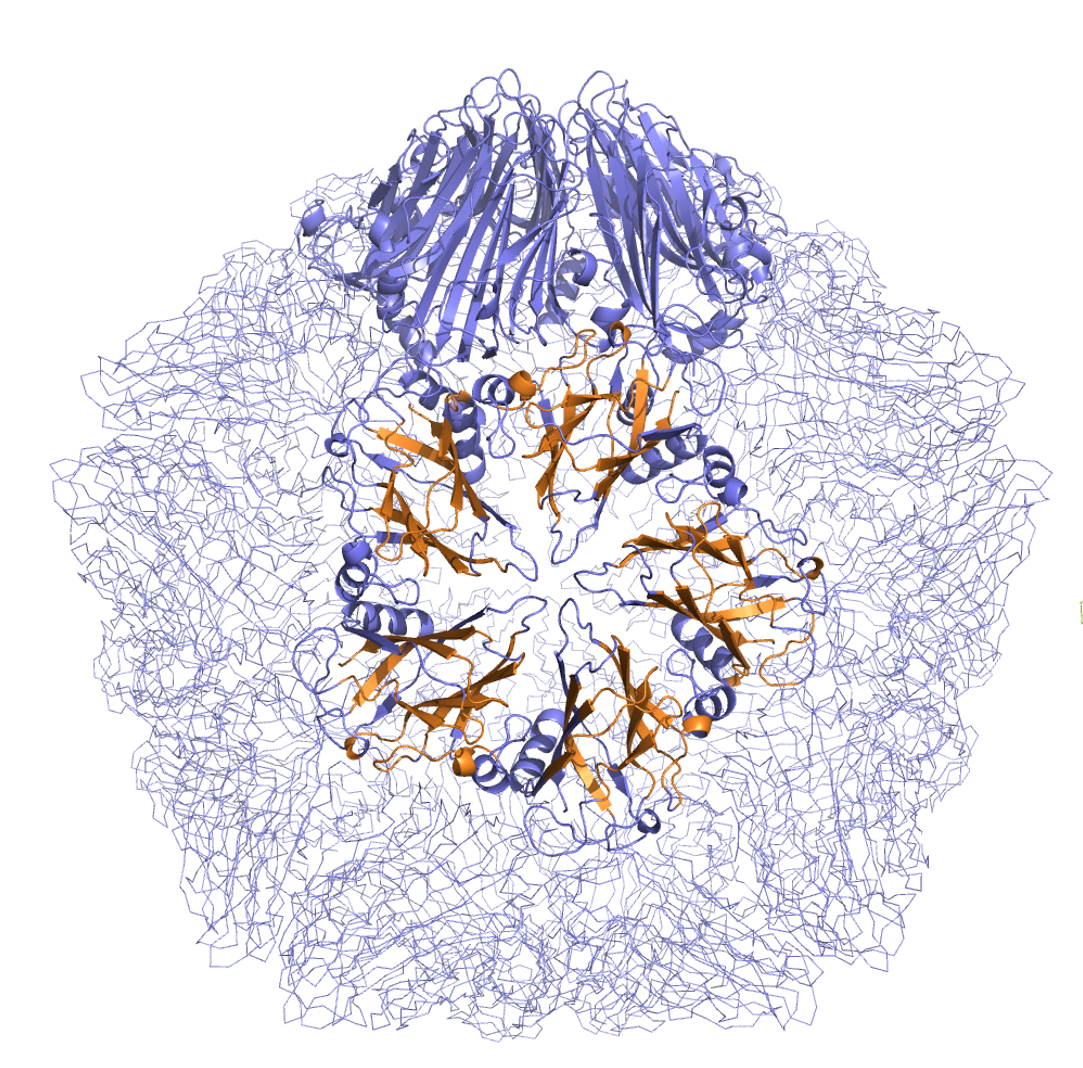
Protein structures are frequently related by spectacular and often surprising similarities. Structural correlations among protein chains are routinely detected by various structure-matching techniques, but the comparison of oligomers and molecular complexes is largely uncharted territory. Here we solve the structure-matching problem for oligomers and large molecular aggregates, including the largest molecular complexes known today. We provide several challenging examples that cannot be handled by conventional structure-matching techniques and we report on a number of remarkable correlations. The examples cover the cell-puncturing device of bacteriophage T4, the secretion system of P. aeruginosa, members of the dehydrogenase family, DNA clamps, ferredoxin iron-storage cages, and virus capsids.
![]() Availability: http://topmatch.services.came.sbg.ac.at
Availability: http://topmatch.services.came.sbg.ac.at
Science ORF Interview
During the Technologiegespräche in Alpbach Prof. Sippl gave an interview on science.orf.at about experimental errors in solved protein structures and bioinformatics. Click here to read the full interview (in german).
COPS Benchmark: interactive analysis of database search methods
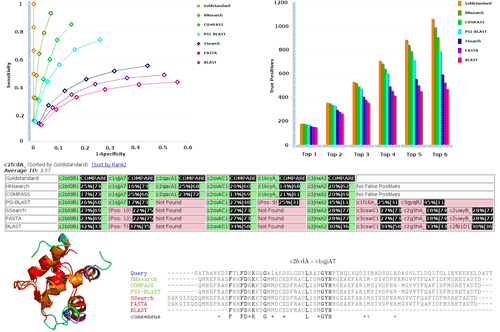
The performance of sequence database search methods is usually judged by receiver operating characteristic (ROC) analysis. The proper interpretation of the results obtained and a fair comparison across different methods critically depends on the properties of the data set used for such an analysis; in particular, each query must have the same number of true positives and true negatives. The COPS-Benchmark is specifically designed for ROC analysis while analysis and results are presented through an intuitive web interface.
The analysis provides details such as false positives per query, and visualization of the structural similarity between query and targets. Most importantly, results obtained for a specific alignment method are immediately related to those obtained for several popular standard sequence alignment methods (BLAST, PSI-BLAST, FASTA, SSearch, COMPASS and HHsearch).
![]() Reference: Bioinformatics 2010 26(4), pp. 574-575
Reference: Bioinformatics 2010 26(4), pp. 574-575
![]() Availability: http://benchmark.services.came.sbg.ac.at
Availability: http://benchmark.services.came.sbg.ac.at
Detection of unrealistic molecular environments in protein structures based on expected electron densities
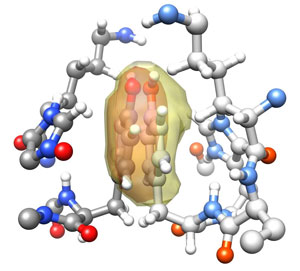
Understanding the relationship between protein structure and biological function is a central theme in structural biology. Advances are severely hampered by errors in experimentally determined protein structures. Detection and correction of such errors is therefore of utmost importance. Electron densities in molecular structures obey certain rules which depend on the molecular environment. RefDens relates electron densities computed from a structural model to densities expected from prior observations on identical or closely related molecular environments. Strong deviations of computed from expected densities reveal unrealistic molecular structures. Most importantly, the electron densities solely depend on the molecular model, so that structure analysis and error detection are independent and hence complementary to experimental data.
![]() Availability: http://refdens.services.came.sbg.ac.at
Availability: http://refdens.services.came.sbg.ac.at
COPS - a novel workbench for explorations in fold space.
Classification of protein structures implies several intricate tasks like the characterization of the classification unit or the definition of the properties to organize these units. But perhaps the most important aspect of any structure classification is that the complete repertoire of available structures is represented in a way that is accessible and comprehensible to consumers who are not necessarily experts in domain decomposition and structure comparison. This requires appropriate user interfaces for navigation in fold space and the instant visualization of structural similarities. The COPS web service (Classification Of Protein Structures) provides an example of current developments in this area.
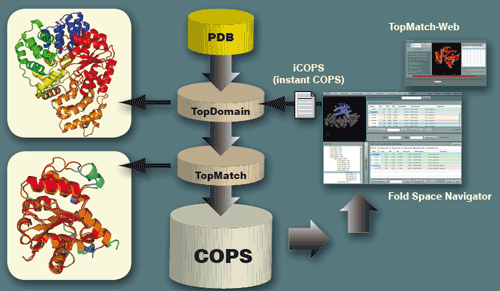
The COPS Hierarchy
Protein domains in COPS are organized as a tree where the domains correspond to tree nodes and pairwise structural similarities among domains correspond to tree edges. The edges represent relative similarities among protein domains derived from structure superpositions. The classification layers of COPS are obtained by cutting the tree at constant relative similarity. Currently, the Fold Space Navigator of COPS displays five layers called distant (30% relative similarity), remote (40%), related (60%), similar (80%) and equivalent (99%).
![]() Download the full poster presentation.
Download the full poster presentation.
![]() Reference: Nucleic Acids Research Vol. 37 (Web Server issue), pp. W539-W544
Reference: Nucleic Acids Research Vol. 37 (Web Server issue), pp. W539-W544